Spongiformsed entsefalopaatiad (prioonhaigused) on need haigused, mille arengus osalevad prioonvalkude patoloogilised vormid. Me teame prioonhaigustest üha enam, kuid põhiaspektid jäävad teadmata - praegu pole meditsiinil vahendeid patsientide ravimiseks nende haiguste vastu.
Spongiformsed entsefalopaatiad ehk prioonhaigused võivad areneda elu jooksul, teised aga pärinevad juba sündides tekkinud pärilikest geenimutatsioonidest. Selles rühmas on inimestel mitu üksust, näiteks Creutzfeldti-Jakobi tõbi või fataalne perekondlik unetus.
Prioonhaigused on pikka aega olnud väga salapärased. Erinevalt teistest patogeenidest, nagu bakterid, viirused või seened, ei sisalda need nukleiinhapet - prioonid on valmistatud ainult valkudest. Prioonhaiguste teooria avastas S. Prusiner, seda avastust hinnati teadlaskonnas väga kõrgelt - 1997. aastal pälvis teadlane Nobeli meditsiinipreemia. Kuigi prioonide kontseptsiooni sünnist on möödunud suhteliselt palju aastaid, usuvad mõned teadlased, et see on siiski puudulik, ja uurivad nende seisundite olemust edasi - mõned spongioossete entsefalopaatiate põhjustajad on nüüdseks kinnitatud.
Prioonhaigused: põhjused
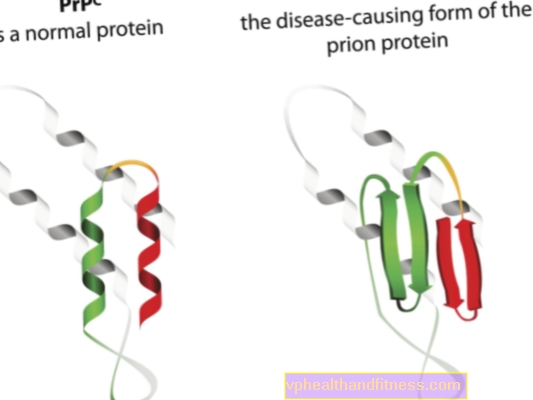
Prioonhaiguste etioloogia on seotud normaalsete prioonvalkude muundamisega patogeenseteks, patogeenseteks vormideks. Prioonid on valgumolekulid, mida leidub iga inimese kehas. Nende funktsioon pole veel päris selge, kuid on teada, et tavatingimustes ei kahjusta prioonvalgud organismi. Kui aga prioonid muudavad oma struktuuri ja muutuvad patogeenseteks osakesteks, tekib üks paljudest spongioossetest entsefalopaatiatest. Organismis looduslikult esinevaid prioone nimetatakse PRPC-ks, ebanormaalseid vorme aga PRPSC-ks. Viimased on tõsine probleem mitte ainult seetõttu, et nad võivad närvikoesse ladestumiste kujul koguneda ja seda kahjustada, vaid ka seetõttu, et neil on võime muundada normaalsed prioonid väärarenguks (lihtsalt öeldes, PRPSC võib "nakatada" normaalseid valke oma patogeense potentsiaaliga).
Loe ka: Huntingtoni tõbi (Huntingtoni korea): põhjused, sümptomid, ravi Lihasvärinad - põhjused. Mida tähendab lihasvärin? Haigused, mis tapavad kõige kiiremini: ŠOKK, EBOLA, DAMN, RÜNNAK, HÄDAOLU [GALE ...Põhimõtteliselt on spongioosse entsefalopaatia 3 põhjust:
- sporaadiline (patogeenset mutatsiooni esineb somaatilistes rakkudes, see toimub patsiendi elu jooksul),
- perekond (tuleneb vanematelt päritud mutatsioonide koormusest),
- Läbipääs (seotud patogeensete prioonide sissetoomisega inimkehasse, näiteks nende osakestega saastunud kasvuhormooni preparaatide või mõne spongiformse entsefalopaatia all kannatava inimese sarvkesta siirdamise kaudu).
Spongiformsed entsefalopaatiad: Creutzfeldt-Jakobi tõbi
Creutzfeldti-Jakobi haigust (CJD) kirjeldati esmakordselt 1920. aastate alguses. Haigust on 4 tüüpi:
- sporaadiline CJD (kõige tavalisem, moodustades kuni 9/10 kõigist CJD juhtudest)
- kodulinn CJD
- CJD-st rabatud
- CJD variant
Creutzfeldt-Jakobi tõve erinevate variantide käigus on kliiniline pilt varieeruv. Selle spongioosse entsefalopaatia rühma kõige levinumad vaevused on:
- dementsuse häired (sealhulgas progresseeruv mälu, tähelepanu ja keskendumisvõime halvenemine)
- müokloonus (tahtmatud liigutused, nagu äkilised lihastõmblused)
- väikeaju düsfunktsioon (avaldub näiteks tasakaalu häiretes)
- ähmane nägemine
- püramiidi- ja ekstrapüramidaalsed sümptomid
CJD variantide käigus võivad ilmneda ka psüühikahäired (nt ärevus, depressiivne meeleolu), valu ja muud tahtmatud liigutused, mida pole eespool nimetatud.
Creutzfeldt-Jakobi tõve prognoos on halb - näiteks juhusliku CJD-ga patsientidel kulub haiguse sümptomite tekkimisest surmani keskmiselt neli kuni viis kuud.
Spongiformsed entsefalopaatiad: Gerstmanni-Straussleri-Scheinkeri sündroom
Gerstmanni-Straussleri-Scheinkeri sündroom (GSS) kulgeb tavaliselt perekondades ja selle põhjustab pärilik mutatsioon PRNP geenis. Seda peetakse kõige aeglasemalt progresseeruvaks spongioosseks entsefalopaatiaks. GSS-i meeskonda kuuluvad:
- spinotserebellaarne ataksia
- düsartria
- dementsuse häired
- neelamishäired
- nüstagm
- suurenenud lihaspinge
Patsientidel, kellel on diagnoositud GSS, on erinev aeg ja mõnel patsiendil toimub surm rohkem kui 10 aastat pärast selle algust.
Spongiformsed entsefalopaatiad: fataalne perekondlik unetus
Surmav perekondlik unetus on prioonhaigus, mis on põhjustatud PRNP geeni mutatsioonidest. Haigus on äärmiselt haruldane ja seda on seni diagnoositud 28 perekonnas kogu maailmas. Surmava perekondliku unetuse käigus on esimene sümptom magamatus. Selle probleemi tagajärjel tekivad ärevushäired ja patsiendil hallutsinatsioonid. Öörahu pideva puudumise tagajärjeks on autonoomse süsteemi toimimise häired (sealhulgas muutused südame töös, higistamine ja seedesüsteemi häired), samuti toimub kehakaalu järkjärguline langus. Fataalse perekondliku unetuse kaugelearenenud staadiumides ilmnevad hormonaalsed häired ja haiguse käigus ilmnevad dementsuse sümptomid.
Surmaga lõppeva perekondliku unetuse, nagu ka teiste spongioossete entsefalopaatiate, prognoos on halb: patsiendid surevad tavaliselt kolme aasta jooksul pärast nende tekkimist.
Spongiformsed entsefalopaatiad: muutuva proteaasitundlikkusega prionopaatia
Käsitletud spongioossete entsefalopaatiate esinemine on peamiselt seotud mutatsioonidega PRNP geenis. Need mutatsioonid puudutavad selle geeni erinevaid koodoneid ja seetõttu eristatakse mitmeid erinevaid prioonhaigusi. Suhteliselt hiljuti kirjeldatud (2008. aastal) üksus on muutuva proteaasitundlikkusega prionopaatia. Selle haiguse all kannatavatel inimestel on mutatsioonid PRNP geeni kolmes koodonis.
Muutuva proteaasitundlikkusega prionopaatia korral saavad patsiendid:
- kognitiivsed häired
- psühhiaatriliste häirete äärmine raskusaste: need võivad olla eufooria ja erutus, aga ka märkimisväärne apaatia
- düsartria
- afaasia (keelehäired)
Haiguse keskmine kestus selles prionopaatias on vähem kui 4 aastat.
Spongioossed entsefalopaatiad: kuru
Kuru peetakse nüüd haiguseks, mida praktiliselt enam ei eksisteeri - see leiti Paapua Uus-Guineast pärit hõimude esindajatelt, kes harrastasid kannibalistlikku käitumist. Selle spongiformse entsefalopaatia domineeriv sümptom on progresseeruv väikeaju ataksia. Sellega võivad kaasneda tahtmatud liigutused (peamiselt korea, värisemise ja atetoosi kujul), samuti kuseteede ja väljaheidete kusepidamatus. Kuri all kannatavad patsiendid kogevad ka olulisi meeleolumuutusi, neil tekivad primitiivsed refleksid (nt imemine). Selle prioonhaiguse puhul on üsna iseloomulik probleem sunnitud nutmine või naermine - viimase nähtuse tõttu nimetatakse kuru mõnikord "naersurmaks".
Spongioossed entsefalopaatiad: diagnoos
Prioonhaigusi võib kahtlustada patsiendi sümptomite põhjal. Need on siiski üsna mittespetsiifilised, kuna võivad ilmneda ka paljude muude haiguste käigus, mis pole seotud prioonidega. Sel põhjusel kasutatakse spongioossete entsefalopaatiate diagnoosimisel ka järgmist:
- pildistamistestid (nt magnetresonantstomograafia, mis võimaldab tuvastada muutusi, mis on seotud aju degeneratsiooniga prioonvalkude poolt),
- laboratoorsed testid (näiteks valgu kontsentratsiooni hindamine tserebrospinaalvedelikus, nt MAP-tau, S-100 või 14-3-3 valgud),
- geneetilised testid (mutatsioonide tuvastamiseks patsiendil),
- immunhistokeemilised testid (antikehade kasutamine prioonvalkude vastu).
Diagnoosi saab kinnitada ka aju lahanguga, mille käigus on võimalik leida spongioossetele entsefalopaatiatele iseloomulikke muutusi. Need võivad olla erineval viisil jaotatud ja erineva struktuuriga (sõltuvalt konkreetsest haigusüksusest) käsnkoldekahjustused, amüloidnaastud ja neuronaalsed defektid.
Spongioossed entsefalopaatiad: ravi
Prioonhaigused on praegu ravimatud - hoolimata paljudest uuringutest, mis on kestnud juba aastaid, pole meditsiinil endiselt ravimeid, mis võiksid nende arengut pidurdada või täielikult pärssida. Spongioosse entsefalopaatiaga patsientidel kasutatakse sümptomaatilist ravi, mille eesmärk on leevendada sümptomite intensiivsust ja parandada nende elukvaliteeti nii palju kui võimalik.
Töö spongioossete entsefalopaatiate ravimisega on aga endiselt pooleli. Teadlased üritavad kasutada erinevaid meetodeid - esimene näide on geeniteraapia. Need mõjutaksid nukleiinhappeid ja nende struktuuris esinevaid mutatsioone - geeniteraapia rakendamise eesmärk oleks vigade neutraliseerimine geneetilises koodis. Teine lähenemisviis on immunoteraapia alus - käimas on antikehade loomine, mille roll oleks patogeensete prioonide kõrvaldamine. Teine meetod, mis näeb ette spongioossete entsefalopaatiate vastu võitlemise võimalusi, on ravi sünteesitud valgu molekulide kasutamisega, mis patsiendi kehasse sattudes neutraliseeriks patoloogilised valgud.
Soovitatav artikkel:
Entsefalopaatiad - põhjused, tüübid ja sümptomid