Cockayne'i sündroom (Neill-Dingwalli sündroom) on haruldane autosoomne retsessiivne mitut süsteemi hõlmav haigus, mille põhjustab molekulaarne defekt, mis kahjustab DNA parandusmehhanismi. Aastane esinemissagedus Euroopa riikides on peaaegu 1/200 000
Cockayne'i sündroomi korral ilmnevad patsiendi rakkudel spetsiifiline defekt geenides, mis on seotud UV-indutseeritud DNA muutuste eemaldamisega aktiivselt transkribeeritud geenides. Sündroomi nahaväliste sümptomite korral võeti arvesse ka täiendavaid defekte põhitranskriptsioonis või oksüdatiivses paranduses.
Haiguse sümptomid ja tüübid
Cockayne'i sündroomi on 3 tüüpi:
- I tüüp (klassikaline) - esialgu ei täheldata kõrvalekaldeid normist, mõnikord võib esineda mikrotsefaaliat, laps võib kehakaalu kehvasti kasvatada. Järk-järgult toimub nägemise ja kuulmise halvenemine, nii kesk- kui ka perifeerse närvisüsteemi degeneratsioon, mis põhjustab enneaegset suremust (elu esimene - teine kümnend)
- II tüüp (tserebro-silma-näo-skeleti sündroom) - kõige raskem, viib elu esimesel kümnendil surma. Seda iseloomustab häiritud neuroloogiline areng. Rasvakude ja aju kaovad, katarakt ja osteoporoos arenevad. KOK-i sündroom on Cockayne'i sündroomi kliinilise spektri äärmuslik sünnieelne vorm, mida iseloomustavad kaasasündinud miinimumid ja artrogrüpoos (polüartikulaarsed kontraktuurid).
- III tüüp - kõige kergemad, sümptomid sarnanevad I tüübiga, kuid vähem rasked. See võimaldab teil jõuda täiskasvanuikka, mõnikord isegi 4-5 aastakümneni.
Sümptomite raskusaste ja haiguse tekkimise vanus varieeruvad sõltuvalt mutatsiooni asukohast. Klassikalise 1. tüüpi Cockayne'i sündroomi korral ilmnevad esimesed sümptomid kõige sagedamini esimesel eluaastal. Samuti on teatatud varasema (sünnieelne) raskemate sümptomitega (II tüüp) ja hilisemate kergemate sümptomitega (III tüüp) juhtudest.
Haiguse kõige levinumad sümptomid on:
- kasvu järkjärguline pidurdamine
- väikeaju ataksia
- spastilisus (üldised ja liigsed lihaste kokkutõmbed, mis takistavad normaalseid liikumisi)
- intellektipuue
- perifeerne sensineuraalne demüeliniseeriv neuropaatia
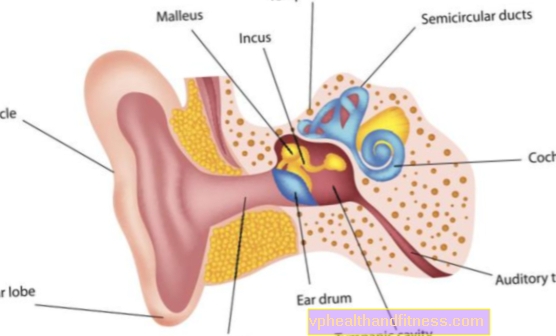
- kuulmislangus
- pigmentaarne retinopaatia
- hambadefektid (kaariese olemasolu)
Tüüpiliste näojoonte hulka kuuluvad mikrotsefaalia, suured kõrvad, kitsas nina ja enoftalmia (silmamuna variseb silmakoopasse, kui orbiidi sisu väheneb).
Mõnel patsiendil on täheldatud katarakti ja valgustundlikkust ning pigmendi retiniiti, mis võib põhjustada pimedaksjäämist.
Samuti täheldati oklusaalset ja neerufunktsiooni häireid, samuti seksuaalse küpsemise puudumist või hilinemist.
Uute mutatsioonide ja vähi oht suureneb.
Seal on nahaaluse rasva kadu, mis võib põhjustada naha enneaegset vananemist.
Diagnoos
A-tüüpi haigus on põhjustatud mutatsioonist ERCC8 geenis kromosoomis 5q11. B tüüp põhjustab mutatsiooni ERCC6 geenis lookuses 10q11.23.
Seda saab tuvastada radioaktiivse testi abil fibroblastikultuurides, et mõõta DNA sünteesi taastumist pärast UV-kiirgust. DNA parandamise test on sündroomi diagnoosimisel otsustav vahend.
Sünnieelne diagnoos
Sünnituseelne diagnoosimine on võimalik kas amniotsüütides või koorioni villides testimisega (samamoodi nagu sündides) või otse molekulaarse järjestuse abil, kui perekonnas on tuvastatud haigust põhjustav mutatsioon.
Ravi
Puudub põhjuslik ravi. Ravi on ainult sümptomaatiline ja hõlmab füsioteraapiat, päikesekaitset, kuuldeaparaate ning sageli tuubisöötmist või gastrostoomiat.